Dystrophies cornéennes
Définition
Une dystrophie cornéenne est une anomalie génétique ou héréditaire, d’une ou plusieurs des couches de la cornée. Sa survenue n’est pas dépendante de facteurs environnementaux, inflammatoires ou systémiques, bien qu’ils puissent parfois influer sur son évolution.
Une dégénérescence cornéenne est une altération qui fait perdre à la cornée une ou plusieurs de ses propriétés normales.
La frontière entre dystrophie et dégénérescence n’est pas toujours franche. En effet, les avancées technologiques et biologiques permettent de décrire des caractéristiques communes familiales pour des atteintes jusque-là réputées dégénératives. Un exemple typique est le kératocône.
Qu’est ce que la cornée?
La cornée est une membrane fibreuse et transparente qui se trouve en avant de l'œil. Avec le cristallin, elle constitue le système optique de l’œil et permet la convergence de la lumière en arrière de l'œil, sur la rétine. La cornée est la lentille (le dioptre) le plus puissant de l’œil (42 dioptries en moyenne).
Elle est composée de 3 couches cellulaires : l’épithélium en superficie, le stroma et l’endothélium en profondeur.
Les différentes dystrophies
Il existe de nombreuses dystrophies cornéennes avec une incidence sur la population qui reste faible. Elles affecteraient 0,09 % de la population et environ 60 % seraient d’origine endothéliale. Pour ne pas faire un long catalogue fastidieux, nous n’évoquerons ici que les dystrophies les plus fréquentes.
Nous ne parlerons pas du kératocône qui bénéficit d’un chapitre à part.
La classification internationale des dystrophies de cornée utilise désormais la génétique moléculaire comme élément de certitude diagnostic. Pour autant, lorsque le phénotype des dystrophies est bien analysé, il est possible d’orienter un diagnostic, de conseiller utilement un patient ou une famille, et d’élaborer une stratégie de prise en charge adaptée.
Dystrophies épithéliales et sous-épithéliales
La dystrophie juvénile héréditaire épithéliale de Meesmann
Elle est autosomique dominante. C'est une atteinte des cytokératines K3 ou K12, qui s’accumulent dans le cytoplasme et la membrane basale épithéliale. C’est une maladie rare de prévalence non connue.
On peut observer une baisse d’acuité visuelle (BAV) modérée et progressive. Des douleurs aiguës ou chroniques de surface oculaire peuvent apparaître en cas de kératopathie ponctuée superficielle. Elle survient dans les premières années de vie et progresse par la suite. Des microvacuoles intra-épithéliales, translucides, punctiformes, milliaires, se forment dans l’aire centro-cornéenne puis se disséminent dans toute l’aire cornéenne. Elles sont localisées préférentiellement dans les couches épithéliales basales et intermédiaires.
On propose habituellement des lubrifiants de surface oculaire. L’ablation de l’épithélium n’est pas efficace sur la symptomatologie en raison de récidive précoce de l’affection. La dystrophie reste souvent asymptomatique. Une douleur ou une BAV peuvent se chroniciser car la récidive est précoce après détersion épithéliale. Le pronostic reste favorable car la symptomatologie est peu handicapante.
L’épithéliopathie microkystique de Cogan
L’épithéliopathie microkystique de Cogan est bien définie mais n’est peut-être pas une dystrophie. La majorité des cas semblent isolés.
Possiblement autosomique dominante, son génotype n’est pas établi. Il s’agit d’une pathologie de la membrane basale de l’épithélium. Elle donne un aspect de cartes géographiques. Les microkystes sont formés par des zones d’épithélium entravées dans son processus de desquamation. Les aspects en empreinte digitale correspondent à l’épithélium mal adhérent qui se plicature et cicatrise.
La prévalence de cette affection sous-diagnostiquée a été estimée entre 2 et 43 % de la population. Cette entité représente 20 % des causes d’érosions épithéliales récidivantes (EER).
Le phénotype de l’épithéliopathie microkystique de Cogan est parfaitement décrit. Il s’exprime habituellement chez l’adulte jeune. On observe l’association d’aires géographiques délimitées par des opacités linéaires, de plicatures épithéliales en empreintes digitales et de vésicules microkystiques intraépithéliales. Elle n’est pas forcément symptomatique. De fait, elle peut être diagnostiquée tardivement. La symptomatologie s’étoffe avec le temps. L’instabilité épithéliale se manifeste par une gêne mécanique. Au stade d’EER, des crises extrêmement douloureuses et brutales s’enchaînent à la moindre friction. À un stade plus avancé, elles réveillent. La succession d’ulcères occasionne une opacité cornéenne, une BAV, une diplopie monoculaire.
Initialement, le traitement est médical. Il utilise des lubrifiants de surface oculaire, des cicatrisants, un mydriatique antalgique et des antalgiques généraux. L’antibiothérapie locale a été proposée sans preuve formelle de sa justification. Les EER avec BAV impose un traitement chirurgical par photokératectomie thérapeutique (PKT). La PKT est efficace dans 95 % des cas. Elle peut être renouvelée. Les microkystes et les cartes géographiques ne semblent pas récidiver. Lorsque la stabilité épithéliale est acquise, les plicatures intraépithéliales en empreinte digitale disparaissent.
Les érosions épithéliales récidivantes (EER) atraumatiques
Plusieurs variantes d’EER dystrophiques existent : la dystrophie de Franceschetti, la dystrophie de Småland (dystrophia Smolandiensis ), la dystrophie d’Hälsingland (dystrophia Helsinglandica ) et la dystrophie cornéenne sous-épithéliale mucineuse. La transmission est autosomique dominante sans gène identifié.
Les érosions récurrentes créent une cicatrisation qui est disposée en pannus sous-épithélial dans la dystrophie de Fanceschetti, localisée dans le tiers cornéen inférieur. Des dépôts de type amyloïdes peuvent être régulièrement observés. On estime que la moitié des EER sont dystrophiques.
L’EER est le signe prédominant, le plus précoce et le plus handicapant et survient parfois dès la naissance. Le phénotype est celui des EER traumatiques mais avec une transmission familiale. S’y associent une fibrose sous-épithéliale progressive avec parfois des bulles intraépithéliales (Franceschetti) ou des petites élevures gélatineuses microscopiques au niveau des couches basales dans les formes évoluées. Des dépôts pseudo-kéloïdiens peuvent être observés au niveau de la couche de Bowman dans la dystrophie de Småland.
Le traitement médical précède le traitement chirurgical (PKT). La PKT traite aussi le pannus fibrotique et peut être renouvelée. La chirurgie est efficace et permet de restaurer une stabilité épithéliale.
La dystrophie de Lisch
Elle est récessive liée à l’X. Le gène et la physiopathologie restent inconnus.
L’incidence est inférieure à 1/1 000 000, probablement sous-estimée, car confondue avec les kératopathies en bandelette. Elle est asymptomatique, occasionnant parfois une BAV ou une photophobie. Une opacité épithéliale microgranuleuse brunâtre opaque s’étend en bandelette dans l’épithélium des cornées atteintes. Elle apparaît chez l’adulte. La progression est lente, généralement de la périphérie vers le centre.
La PKT traite une BAV lorsqu’elle est symptomatique et gêne le patient. Il faut éliminer un carcinome in situ. Elle est de bon pronostic.
La dystrophies liées au gène TGFβI
Il s’agit d’une famille de dystrophies liée à différentes mutations du gène transforming growth beta factor induced protein H3 (TGFβ I). Il code pour la kératoépithéline. La protéine mutée qui s’accumule exclusivement dans la cornée donne un phénotype spécifique à sa mutation. Toutes les atteintes de ce gène sont autosomiques dominantes, même si la dystrophie grillagée de type 3A est d’expressivité et de pénétrance variable, et ressemble à une forme récessive.
La dystrophie granulaire de type I classique, et sa variante superficielle, sont les plus courantes. Elles sont retrouvées chez la moitié des familles atteintes. Faute de recensement exhaustif et non redondant des cas avérés et génotypés, la prévalence de ces affections rares n’est pas clairement établie. Elle avoisinerait les 137 pour 1 000 000 personnes.
Les phénotypes homozygotes sont plus marqués et plus denses que les hétérozygotes. Les atteintes cornéennes épargnent généralement la région pré-limbique. La mutation est initialement asymptomatique puis entraîne une photophobie et/ou EER, puis une BAV et des contrastes.
On distingue habituellement :
- les atteintes de la couche de Bowman et du stroma antérieur principalement avec les dystrophies de Reis-Bücklers ou de Thiel-Behnke et leurs variantes
- les atteintes du stroma, principalement avec les dystrophies granulaires type 1 (la plus fréquente), granulaires type 2 (Avellino), grillagées type 1 (Biber-Haab-Dimmer et variantes : début précoce) ou grillagées de type 3 (et 3A) à début tardif et grillagées type 4 (japonaises).
Le traitement est motivé par les signes fonctionnels. Il fait appel à la PKT pour stabiliser l’épithélium ou pour photo-ablater les opacités. Les opacités invalidantes profondes indiquent une kératoplastie lamellaire antérieure (KLA).
Les Dystrophies stromales non liées à TGFβI
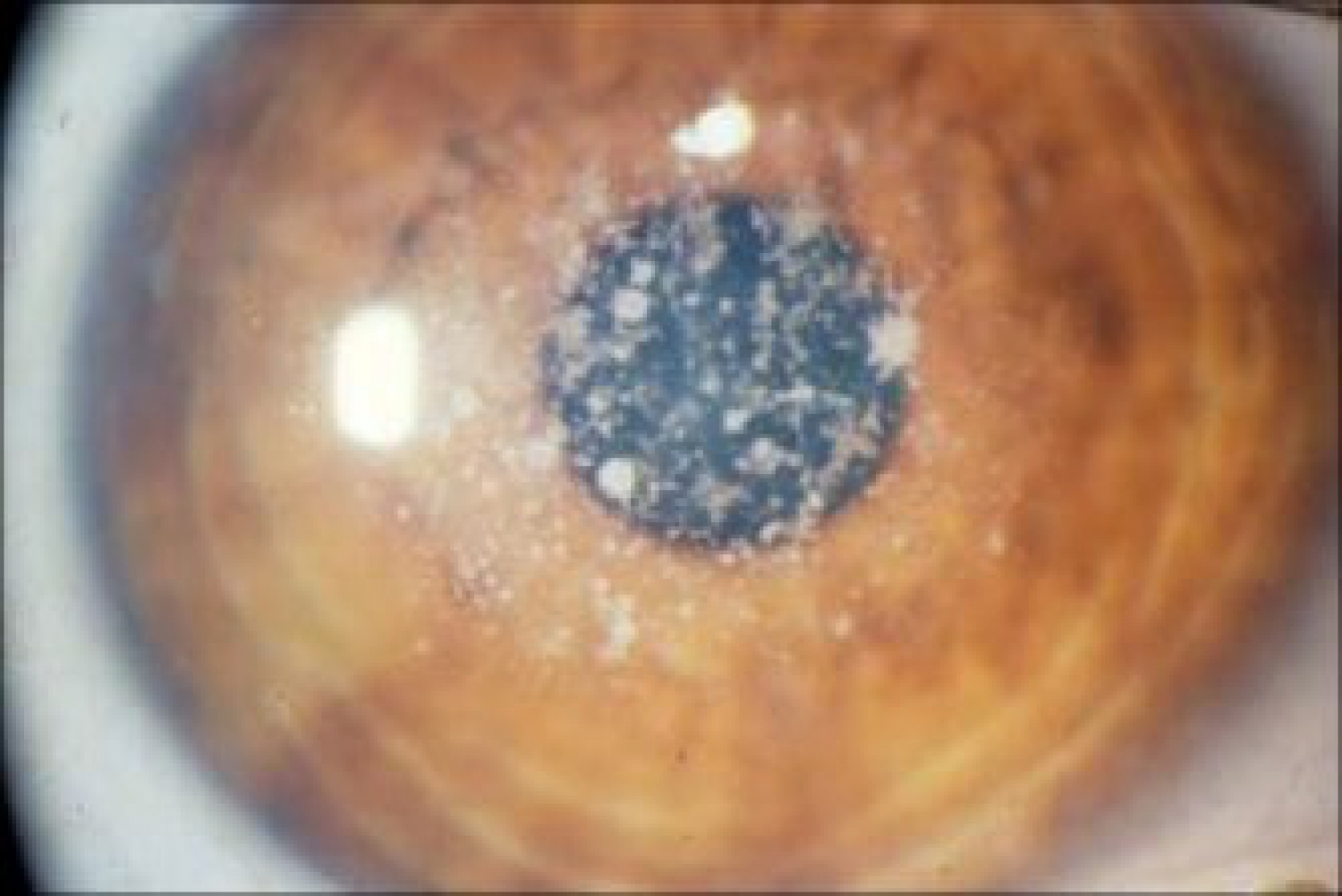
La dystrophie granulaire maculaire (anciennement Groenouw 2)
Autosomique récessive, elle entraîne l’accumulation de dépôts de kératane sulfate dans toutes les couches cornéennes. On en distingue trois types selon la réactivité antigénique. Sa prévalence est rare, estimée à 2 pour 100 000.
Le phénotype se déclare dans la première décade et progresse sur plusieurs années. Les dépôts s’arrangent en macules fibrogranulaires blancs-grisâtres, à bords flous (en neige sale), dans toutes les couches, sur toute l’aire cornéenne. La sensibilité cornéenne est diminuée, parfois absente. Ces dépôts sont denses et réflectifs, obturants. La baisse d’acuité prime avec la photophobie. Les EER (érosions épithéliales récidivantes) sont possibles. Les explorations sont communes aux autres dystrophies, avec un dosage sanguin du kératane sulfate qui précise le type dystrophique.
La PKT est rarement indiquée, sauf en cas d’instabilité épithéliale douloureuse. Les dépôts profonds du stroma obligent à la kératoplastie pour traiter la BAV et la photophobie. La kératoplastie privilégie les techniques lamellaires. L’endothélio-descemet étant infiltrée, il faut parfois pratiquer une greffe de cornée.
Autres dystrophies :
- dystrophie cristalline de Schnyder
- dystrophie cornéenne stromale héréditaire (Turpin)
- dystrophie cornéenne mouchetée de François-Neetens
- dystrophie grillagée type 2… etc…
Dystrophies endothélio-descemétiques
La dystrophie de Fuchs ou cornea guttata familiale
Autosomique dominante, sa pénétrance est de 100 % mais d’expressivité variable. L’affection semble en relation avec un dysfonctionnement dans la production du collagène 8 et/ou un facteur de transition métaplasique des cellules endothéliales. Des altérations de l’ADN mitochondrial liées au stress oxydatif ont aussi été incriminées. Le métabolisme des cellules endothéliales pathologiques régresse. Elles produisent à nouveau du collagène 8 embryonnaire qui s’accumule en verrucosités à la face interne de la membrane de Descemet, qui prennent un aspect en goutte (cornea guttata). Au cours de l’évolution, les cellules les plus pathologiques disparaissent et ne sont pas régénérées. À son terme évolutif, l’incompétence fonctionnelle des cellules endothéliales et leur rupture de confluence engendrent un œdème cornéen, initialement au dépend du stroma postérieur central. Lorsque l’œdème augmente, il diffuse dans le reste du stroma, jusque sous l’épithélium qu’il ne peut franchir. Des bulles sous-épithéliales se forment alors et stimulent les terminaisons nerveuses, créant gêne, voire douleur. Les bulles peuvent se rompre au stade de la kératopathie bulleuse.
L’incidence de la dystrophie de Fuchs est supérieure à 1/2000 avec une prédominance féminine. La prévalence serait d’environ 4 à 11 % des individus de plus de 40 ans. Les défaillances endothéliales séniles, avec ou sans cornea guttata , sont plus fréquentes sans transmission familiale et plus tardive.
Le bilan et le suivi utilisent l’OCT de cornée ou la topographie cornéenne pour l’œdème stromal (mieux que la pachymétrie ultrasonore), et surtout la microscopie spéculaire qui explore l’endothélium.
Le traitement, au stade d’œdème débutant, utilise des collyres hyperosmolaires associés à la cortisone locale. Ensuite, la greffe endothéliale s’impose en privilégiant une DSAEK ou une DMEK.
L’évolution est lente, sur 10 à 20 ans. Un déverrouillage visuel matinal inférieur à une demi-heure peut perdurer plusieurs années. L’âge auquel la symptomatologie fonctionnelle de la dystrophie de Fuchs apparaît coïncider généralement avec celui des signes fonctionnels de la cataracte. Le bénéfice d’une phacoexérèse doit donc être envisagée en tenant compte du risque opératoire de perte cellulaire endothéliale accrue.
Population concernée
Elle diffère en fonction de l’étiologie de la dystrophie cornéenne concernée.
Examens nécessaires pour détecter une dystrophie cornéenne
Le biomicroscope
Le biomicroscope (ou lampe à fente) est l’outil utilisé en routine par l’ophtalmologiste pour examiner l’œil de ses patients. Il permet la recherche de signes évocateurs d’une dystrophie cornéenne, de réaliser des clichés photographiques, qui permettront de surveiller l’évolution.
La topographie cornéenne
Elle est obtenue par un topographe cornéen spéculaire dont la principale fonction est de mesurer la forme « topographique » de la face antérieure de la cornée. Son relevé se lit comme une carte géographique (les couleurs chaudes correspondent à des surélévations, les couleurs froides à des dépressions).
Certains topographes sont capables de donner une image topographique des deux faces (antérieure et postérieure) de la cornée : il s’agit de topographes d’élévation.
Une carte différentielle donne ainsi le relevé de l’épaisseur de la cornée en tout point (pachymètrie) et d’appréhender la courbure et la puissance réfractive cornéenne pour adapter la thérapeutique. Les valeurs d’élévation postérieure sont faussées lorsque la transparence cornéenne est fortement altérée.
La pachymétrie
Elle consiste en la mesure de l’épaisseur de la cornée notée en microns (μ). Pour information, une cornée normale mesure environ 12 mm de diamètre, sa pachymétrie est de 520 μ (un demi millimètre) au centre et d’environ 700 μ en périphérie.
La mesure de l’épaisseur de la cornée peut être obtenue de différentes manières :
- par une sonde à ultrasons avec contact de la cornée (principe de l’échographie)
- par topographie cornéenne d’élévation (mesure optique)
- par interférométrie (OCT)
- par microscopie dite confocale (HRT) ou spéculaire
La microscopie spéculaire
La microscopie spéculaire est la technique de référence pour l'exploration de l'endothélium cornéen. Elle permet l'analyse qualitative et quantitative à titre diagnostique, prédictif ou dans un but de simple surveillance, de cette couche monocellulaire dont le rôle dans est le maintien de la transparence cornéenne.
L’OCT du segment antérieur
L’OCT permet d visualiser la localisation, la forme, l’étendue et la profondeur des dépôts ou les anomalies cornéennes dystrophiques.
Traitement
Les principes thérapeutiques
Les traitements ne peuvent actuellement pas traiter la cause génétique des affections. Ils s’attachent donc à soulager les signes fonctionnels selon la dystrophie. Ils permettent de traiter les érosions épithéliales récidivantes, la baisse d’acuité visuelle, la photophobie, l’œdème ou la douleur.
En fonction des dystrophies, un conseil génétique est parfois utile.
Les traitements médicaux (cicatrisants, lubrifiants, agents hyperosmotiques, lentilles pansements) précèdent les traitements chirurgicaux du moins invasifs (photokératectomie, kératectomie lamellaire) aux plus invasifs (kératoplastie). Selon la dystrophie, une prise en charge pluridisciplinaire peut être nécessaire.
Les greffes de cornée
Le principe de la greffe de cornée est simple. Il s’agit d’échanger la partie centrale de la cornée malade par la même partie de la cornée saine d’un donneur décédé. Le prélèvement, la conservation et la délivrance des greffons sont très strictement codifiés. La greffe de cornée est un geste chirurgical à présent bien maîtrisé. Les kératocônes représentent environ 1/3 des indications de greffes de cornées. Seuls les patients dont l’acuité visuelle est insuffisante avec des lentilles de contact sont inscrits sur la liste d’attente pour un greffon cornéen.
La première étape est l’inscription sur la liste des patients à greffer. L’attente dépend du nombre de cornées prélevées et de la demande pouvant varier considérablement d’un centre à l’autre.
Le jour de l’intervention, le patient arrive à jeun. Le geste chirurgical dure entre 1 heure et 1h30 selon la technique. Une ou deux nuits d’hospitalisation sont en général nécessaires. Un arrêt de travail d’un mois est souvent prescrit. Un traitement antibiotique et anti-inflammatoire est prescrit en collyres à dose dégressive pour une durée de 6 à 12 mois selon la technique.
Il existe plusieurs variantes chirurgicales : On parle de greffe transfixiante lorsque toute l’épaisseur de la cornée est remplacée, de greffe lamellaire antérieure lorsqu’il s’agit uniquement de la partie externe de la cornée qui est greffée et l’on parle de greffe lamellaire antérieure profonde (ou pré-descemétique) lorsque toute la cornée est greffée sauf sa couche la plus postérieure (membrane de Descemet + couche endothéliale). L’endothélium est un élément important de la cornée qui assure la transparence du greffon grâce à un contingent de cellules qui ne peuvent se renouveler.
Les greffes lamellaires antérieures donnent des résultats optiques peu satisfaisants et la greffe pré-descemétique est plus délicate à réaliser. Selon les cas, une greffe pré-descemétique n’est pas réalisable et sera convertie en greffe transfixiante.
Le chirurgien choisira le type de greffe le plus adapté pour chaque patient.
Si les deux yeux sont à greffer, l’opération du deuxième œil n’est théoriquement possible qu’un an après celle du premier. Il en est de même en cas de rejet de greffe.
Les résultats visuels sont globalement bons mais la rapidité de la réhabilitation visuelle est lente. Il faut compter 3 mois avant de retrouver une cornée transparente et au moins 6 mois voire 1 an, avant de pouvoir débuter l’ablation sélective des points de suture. Cette étape permet d’ajuster l’astigmatisme et d’équiper le sujet soit en verres de lunettes soit en lentilles de contact à nouveau.
Les informations délivrées ci-dessus sont données à titre purement informatif.En raison de l'évolution des spécificités de chaque cas, les informations délivrées ne seraient se substituer à celles qui vous seront délivrées en consultation.L’équipe médicale du Centre d’ophtalmologie Jean Jaurès.
